| Course |
· Exposure to inhaled noxious particles and gases causes inflammation of the lungs that can lead to COPD if the normal protective and/or repair mechanisms are overwhelmed or defective.
· Exacerbations of COPD are associated with an increase in airway inflammation.
· Although inflammation is important in both diseases, the inflammatory response in COPD is markedly different from that in asthma.
· In addition to inflammation, two other processes thought to be important in the pathogenesis of COPD are an imbalance of proteinases and anti-proteinases in the lung, and oxidative stress.
· Pathological changes characteristic of COPD are found in the central airways, peripheral airways, lung parenchyma, and pulmonary vasculature.
· The peripheral airways become the major site of airways obstruction in COPD. The structural changes in the airway wall are the most important cause of the increase in peripheral airways resistance in COPD. Inflammatory changes such as airway edema and mucus hypersecretion also contribute to airway narrowing.
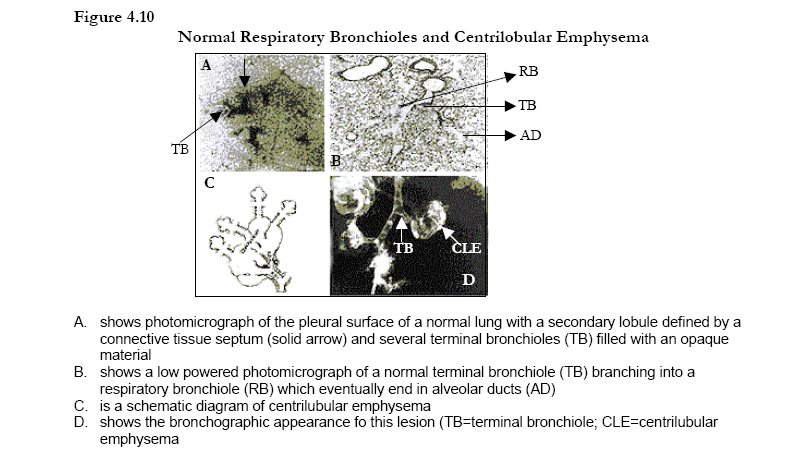
· Most common in COPD patients is the centrilobular form of emphysema, which involves dilatation and destruction of the respiratory bronchioles.
· Physiological changes characteristic of the disease include mucus hypersecretion, ciliary dysfunction, airflow limitation, pulmonary hyperinflation, gas exchange abnormalities, pulmonary hypertension, and cor pulmonale, and they usually develop in this order over the course of the disease.
· The irreversible component of airflow limitation is primarily due to remodeling of the small airways. Parenchymal destruction (emphysema) also contributes but plays a smaller role.
· In advanced COPD, peripheral airways obstruction, parenchymal destruction, and pulmonary vascular abnormalities reduce the lung’s capacity for gas exchange, producing hypoxemia and later on, hypercapnia. Inequality in the ventilation/perfusion ration (VA/Q) is the major mechanism behind hypoxemia in COPD.
· Pulmonary hypertension develops late in the course of COPD. It is the major cardiovascular complication of COPD and is associated with a poor prognosis.
· COPD is associated with systemic inflammation and skeletal muscle dysfunction that may contribute to limitation of exercise capacity and decline of health status.
Inhaled noxious particles and gases that lead to COPD cause lung inflammation, induce tissue destruction, impair the defense mechanisms that serve to limit the destruction, and disrupt the repair mechanisms that may be able to restore tissue structure in the face of some injuries. The results of lung tissue damage are mucus hypersecretion, airway narrowing and fibrosis, destruction of the parenchyma (emphysema), and vascular changes. In turn, these pathological changes lead to airflow limitation and the other physiological abnormalities characteristic of COPD.
Much of the information concerning the pathogenesis of COPD comes from studies in experimental animals or in vitro systems. These experimental systems are limited as they differ from human disease in a number of respects. Studies in human subjects of the pathogenesis, pathology, and pathophysiology of COPD are often limited by patient selection, small numbers of subjects, and limited access to the relevant tissue. Therefore, an evidence-based perspective on these topics is in many respects incomplete.
COPD is characterized by chronic inflammation throughout the airways, parenchyma, and pulmonary vasculature. The intensity and cellular and molecular characteristics of the inflammation vary as the disease progresses. Over time, inflammation damages the lungs and leads to the pathologic changes characteristic of COPD.
In addition to inflammation, two other processes thought to be important in the pathogenesis of COPD are an imbalance of proteinases and anti-proteinases in the lung, and oxidative stress. These processes may themselves be consequences of inflammation, or they may arise from environmental (e.g., oxidant compounds in cigarette smoke) or genetic (e.g., alpha-1 antitrypsin deficiency) factors. Figure 4-1 details the interactions between these mechanisms. The multiplicity of cells and mediators thought to be involved in the pathogenesis of COPD is presented schematically in Figure 4-2.
Figure 4.1 Pathogenesis of COPD

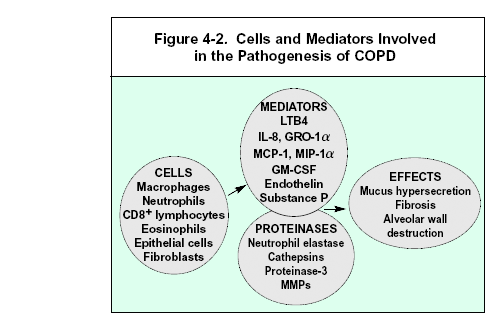 |
COPD is characterized by an increase in neutrophils, macrophages, and T lymphocytes (especially CD8+) in various parts of the lung (Figure 4-3). There may also be an increase in eosinophils in some patients, particularly during exacerbations. These increases are brought about by increases in inflammatory cell recruitment, survival, and/or activation. Many studies reveal a correlation between the number of inflammatory cells of various types in the lung and the severity of COPD1-10.
Figure 4.3 Sites of Inflammatory Cell Increases in COPD

Neutrophils. Increased numbers of activated neutrophils are found in sputum and bronchoalveolar lavage (BAL) fluid of patients with COPD4,5,8,9, although the role of neutrophils in COPD is not yet clear. Neutrophils are also increased in smokers without COPD11. However, neutrophils are little increased in airway and parenchyma tissue sections, which may reflect their rapid transit through these parts of the lung. Induced sputum studies also show an increase in myeloperoxidase (MPO) and human neutrophil lipocalin, indicating neutrophil activation12. Acute exacerbations of COPD are characterized by a marked increase in the number of neutrophils in BAL fluid13. Neutrophils secrete several proteinases, including neutrophil elastase (NE), neutrophil cathepsin G, and neutrophil proteinase-3, which may contribute to parenchymal destruction and chronic mucus hypersecretion.
Macrophages. Increased numbers of macrophages are present in the large and small airways and lung parenchyma of patients with COPD, as reflected in histopathology, BAL, bronchial biopsy, and induced sputum studies2,4-9. In patients with emphysema, macrophages are localized to sites of alveolar wall destruction1. Macrophages likely play an orchestrating role in COPD inflammation by releasing mediators such as tumor necrosis factor-a (TNF-a), interleukin 8 (IL-8), and leukotriene B4 (LTB4), which promote neutrophilic inflammation.
T lymphocytes. Histopathology and bronchial biopsy studies show an increase in T lymphocytes, especially CD8+ (cytotoxic) cells, throughout the lungs of patients with COPD1,2,10,14. Their role in COPD inflammation is not yet fully understood, but one way that CD8+ cells may contribute to COPD is by releasing perforin, granzyme-B, and TNF-a, which can cause the cytolysis and apoptosis of alveolar epithelial cells15 that may be responsible for the persistence of inflammation. An increased number of lymphocyte-like natural killer (NK) cells has also been reported in patients with severe COPD3.
Eosinophils. The presence and role of eosinophils in COPD are uncertain. Some bronchial biopsy studies show eosinophils increased in the airways of some patients with stable COPD6,16. However, some of these patients may have had coexisting asthma, as other studies report no increase in eosinophils in COPD patients2. The levels of eosinophil cationic protein (ECP) and eosinophil peroxidase (EPO) in induced sputum are elevated in COPD, suggesting that eosinophils may be present but degranulated, and therefore no longer recognizable by light microscopy12. The high levels of neutrophil elastase (NE) often found in COPD may be responsible for this degranulation17. Most studies agree that airway eosinophils are increased during acute exacerbations of COPD18,19.
Epithelial cells. Airway and alveolar epithelial cells are likely to be important sources of inflammatory mediators in COPD, though their role in inflammation in this disease has not yet been thoroughly studied. Exposure of nasal or bronchial epithelial cells from healthy volunteers to nitrogen dioxide (NO2), ozone (O3), and diesel exhaust particles results in significant synthesis and release of pro-inflammatory mediators, including eicosanoids, cytokines, and adhesion molecules20. The adhesion molecule E-selectin, involved in recruitment and adhesion of neutrophils, is up-regulated on airway epithelial cells in COPD patients21. Cultured human bronchial epithelial cells from COPD patients release lower levels of inflammatory mediators such as TNF-a and IL-8 than similar preparations from nonsmokers or smokers without COPD, suggesting that some form of down-regulation of inflammatory mediator release may occur in epithelial cells of individuals with COPD20.
Activated inflammatory cells in COPD release a variety of mediators, including a spectrum of potent proteinases22,23, oxidants24, and toxic peptides25. Many of the mediators thought to be important in the disease — notably LTB426, IL-84,7,27, and TNF-a4,16 — are capable of damaging lung structures and/or sustaining neutrophilic inflammation. The damage induced by these moieties may further potentiate inflammation by releasing chemotactic peptides from the extracellular matrix28. Little is yet known about the specific role of these inflammatory mediators in COPD. Studies of the therapeutic use of selective mediator antagonists should identify the molecules relevant in COPD.
Leukotriene B4 (LTB4). LTB4, a potent chemoattractant of neutrophils, is found at increased levels in the sputum of patients with COPD26. It is probably derived from alveolar macrophages, which secrete more LTB4 in patients with COPD. Several potent LTB4 receptor antagonists have been developed for clinical studies and should elucidate further the role of this mediator in COPD. So far there is no evidence that cysteinyl leukotrienes (LTC4, LTD4, LTE4) are involved in COPD. Selective antagonists of the cysteinyl leukotriene 1 receptor (CysLT1) have proven helpful in patients with asthma and studies of these drugs in COPD patients are now underway. The role of the cysteinyl leukotriene 2 receptor (CysLT2) in respiratory disease is as yet unknown29.
Interleukin 8 (IL-8). IL-8, a selective chemoattractant of neutrophils that may be secreted by macrophages, neutrophils, and airway epithelial cells, is present at high concentrations in induced sputum and BAL fluid of patients with COPD,4,7,27. IL-8 may play a primary role in the activation of both neutrophils and eosinophils in the airways of COPD patients and may serve as a marker in evaluating the severity of airway inflammation27.
Tumor necrosis factor-a (TNF-a). TNF-xx activates the transcription factor nuclear factor-kB (NF-kB), which in turn activates the IL-8 gene in epithelial cells and macrophages (Figure 4-4). TNF-a is present at high concentrations in sputum4 and is detectable in bronchial biopsies16 in patients with COPD. TNF-a serum levels and production by peripheral blood monocytes are increased in weight-losing COPD patients, suggesting that this mediator may play a role in the cachexia of severe COPD30.
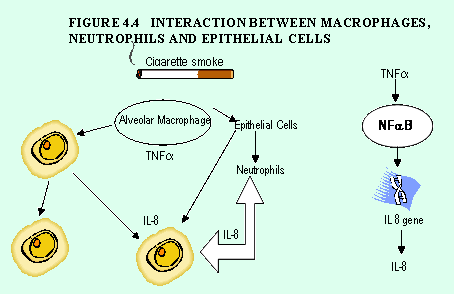
Cigarette smoke activates macrophages and epithelial cells to produce tumor necrosis factor a (TNF-a), switching on the gene for interleukin 8 (IL-8), which recruits and activates neutrophils. This process occurs via activation of the transcription factor nuclear factor - kB (NF-kB).
Others. Other inflammatory mediators that may be involved in COPD include the following:
· Macrophage chemotactic protein-1 (MCP-1), a potent chemoattractant of monocytes, is increased in the BAL fluid of patients with COPD and smokers without COPD, but not in ex-smokers or nonsmokers31. Thus, MCP-1 may be involved in macrophage recruitment into the lungs in smokers.
· Macrophage inflammatory protein-1ß (MIP-1ß) is increased in the BAL fluid of patients with COPD compared to smokers, ex-smokers, and nonsmokers31. Macrophage inflammatory protein-1a (MIP-1a) shows increased expression in airway epithelial cells from COPD patients3 compared to control smokers.
· Granulocyte-macrophage colony stimulating factor (GM-CSF) is found at increased concentrations in the BAL fluid of patients with stable COPD and at markedly elevated levels during exacerbations13. The number of GM-CSF-immunoreactive macrophages is also increased in sputum of patients with COPD32. GM-CSF is important for neutrophil survival and may play a role in enhancing neutrophilic inflammation.
· Transforming growth factor-ß (TGF-ß) and epidermal growth factor (EGF) show increased expression in epithelial cells and submucosal cells (eosinophils and fibroblasts) in COPD patients33. These mediators may play a role in airway remodeling (fibrosis and narrowing) in COPD34.
· Endothelin-1 (ET-1), a potent endothelium-derived vasoconstrictor peptide, is found at increased concentrations in induced sputum of patients with COPD35. Patients with severe COPD also have elevated plasma levels of ET-1, which is probably related to their chronic hypoxemia36.
· Neuropeptides, such as substance P, calcitonin gene-related peptide, and vasoactive intestinal peptide (VIP), have potent effects on vascular function and mucus secretion. An increased concentration of substance P is found in sputum of patients with chronic bronchitis37. One bronchial biopsy study showed an increase in VIP-immunoreactive nerves in the vicinity of submucosal glands in patients with chronic bronchitis, suggesting that this substance may play a role in mucus hypersecretion38. However, another study showed no significant differences in the number of nerves immunoreactive for substance P, calcitonin gene-related peptide, or VIP between COPD patients and healthy subjects39.
· Complement. Activation of the complement pathway via generation of the potent chemotaxin C5a may play a significant role in the neutrophil accumulation seen in the lungs of patients with COPD40.
Although inflammation is important in both diseases, the inflammatory response in COPD is markedly different from that in asthma, as summarized in Figure 4-5. However, some patients with COPD also have asthma, and the inflammation in their lungs may show characteristics of both diseases.
Figure 4.5 Characteristics of Inflammation in COPD and Asthma
|
|
COPD |
Asthma |
|
Cells |
· Neutrophils · Large increases in macrophages · Increase in CD8+ lymphocytes |
· Eosinophils · Small increase in macrophages · Increase in CD4+ Th2 lymphocytes · Activation of mast cells |
|
Mediators |
· LTB4 · IL-8 · TNF-a |
· LTD4 · IL-4, IL-5 · Plus many others |
|
Consequences |
· Squamous metaplasia of epithelium · Parenchymal destruction · Mucus metaplasia · Glandular enlargement |
· Fragile epithelium · Thickening of basement membrane · Mucus metaplasia · Glandular enlargement |
|
Response to treatment |
· Glucocorticosteroids have little or no effect |
· Glucocorticosteroids inhibit inflammation |
Since inflammation is a feature of COPD, it follows that anti-inflammatory therapies may have clinical benefit in controlling symptoms, preventing exacerbations, and slowing the progression of the disease. However, the inflammatory response in COPD appears to be poorly responsive to the glucocorticosteroids that are effective anti-inflammatory medications in asthma.
The connection between cigarette smoke and inflammation has been most extensively studied41-52. Cigarette smoke activates macrophages and epithelial cells to produce TNF-a and may also cause macrophages to release other inflammatory mediators, including IL-8 and LTB453,54.
Inflammation is present in the lungs of smokers without a diagnosis of COPD. This inflammation is similar to, but less intense than, the inflammation in the lungs of patients with COPD. For example, induced sputum studies show that smokers without COPD have a greater proportion of neutrophils in their lungs than age-matched nonsmokers, but a smaller proportion than COPD patients4,9. Thus, the inflammation characteristic of COPD is thought to represent an exaggeration of a normal, protective response to inhalational exposures.
However, not all smokers develop COPD, and why the normal, protective inflammatory response becomes an exaggerated, harmful one in some smokers is poorly understood. Presumably the inflammation caused by cigarette smoking interacts with other host or environmental factors to produce the excess decline in lung function that results in COPD55. Inflammatory changes are also present in bronchial biopsies in ex-smokers, suggesting that the inflammatory response in COPD may persist even in the absence of continuous exposure to risk factors56.
A number of studies have demonstrated that a variety of particulates (e.g., diesel exhaust, grain dust) can initiate respiratory tract inflammation57-61. It is likely that indoor air pollution derived from the burning of biomass fuels will prove to have similar effects.
Laurell and Eriksson observed in 1963 that individuals with a hereditary deficiency of the serum protein alpha-1 antitrypsin, which inhibits a number of serine proteinases such as neutrophil elastase, are at increased risk of developing emphysema62. Elastin, the target of neutrophil elastase, is a major component of alveolar walls, and elastin fragments may perpetuate inflammation by acting as potent chemotactic agents for macrophages and neutrophils. These observations led to the hypothesis that an imbalance between proteinases and endogenous antiproteinases results in lung destruction.
Based on many observations, it now seems clear that an imbalance of proteinases and antiproteinases may involve either increased production or activity of proteinases, or inactivation or reduced production of antiproteinases. Often, the imbalance is a consequence of the inflammation induced by inhalational exposures. For example, macrophages, neutrophils, and airway epithelial cells release a combination of proteinases. The imbalance may also be caused by a decrease of antiproteinase activity by oxidative stress (itself a consequence of inflammation), cigarette smoke63,64, and possibly other COPD risk factors.
The concept has also been expanded to include additional proteinases and antiproteinases. While neutrophil elastase is likely to be the major proteinase involved in lung destruction in alpha-1 antitrypsin deficiency, it may not be involved in COPD caused by inhalational exposures. Additional proteinases that have been implicated in COPD include neutrophil cathepsin G, neutrophil proteinase-3, cathepsins released from macrophages (specifically cathepsins B, L, and S), and various matrix metalloproteinases (MMPs)65. These proteinases are capable of degrading elastin and also collagen, another main component of alveolar walls. Some proteinases, such as neutrophil elastase66 and neutrophil proteinase-367, induce mucus secretion, and neutrophil elastase also produces mucus gland hyperplasia68. Thus, proteinases may be involved in mucus hypersecretion as well as parenchymal destruction. Antiproteinases thought to be involved in COPD include, in addition to alpha-1 antitrypsin, secretory leukoproteinase inhibitor (SLPI) and tissue inhibitors of MMPs (TIMPs).
There is increasing evidence that an
oxidant/antioxidant imbalance, in favor of oxidants, occurs in COPD. (The
process is summarized in Figure 4-6.) Markers of oxidative stress have
been found in the epithelial lining fluid, breath, and urine of cigarette
smokers and patients with COPD. For example, hydrogen peroxide (H2O2)
and nitric oxide (NO) are direct measures of oxidants generated by cigarette
smoking or released from inflammatory leukocytes and epithelial cells. H2O2
is increased in the breath of patients with stable COPD and during acute
exacerbations69, and NO is increased in the breath during
exacerbations of COPD70. A prostaglandin isomer, isoprostane F2a-III,
which is formed by free radical peroxidation of arachidonic acid and believed
to be an in vivo biomarker of lung oxidative stress, is increased in
both breath condensates71 and urine72 in COPD patients
compared to healthy controls and is increased even more during exacerbations.
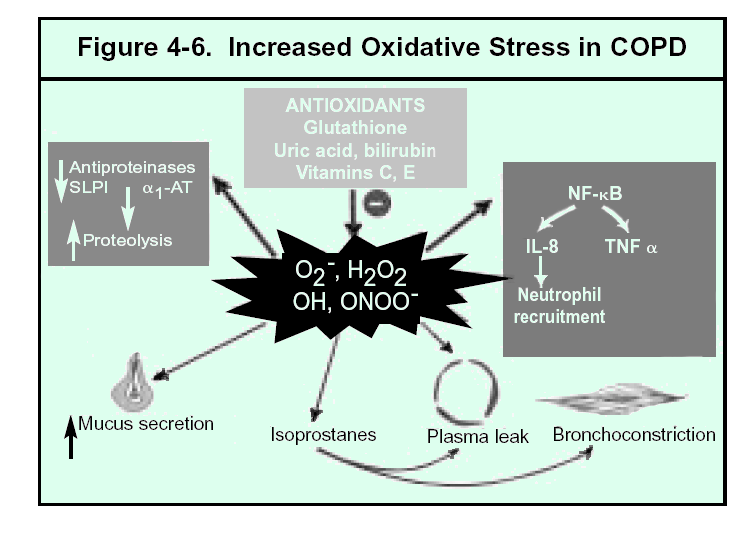
Oxidative stress contributes to COPD in a variety of ways. Oxidants can react with, and damage, a variety of biological molecules, including proteins, lipids, and nucleic acids, and this can lead to cell dysfunction or death, as well as damage to the lung extracellular matrix. In addition to directly damaging the lung, oxidative stress contributes to the proteinase-antiproteinase imbalance both by inactivating antiproteinases (such as alpha-1 antitrypsin and SLPI) and by activating proteinases (such as MMPs). Oxidants also promote inflammation, for example by activating the transcription factor NF-kB, which orchestrates the expression of multiple inflammatory genes thought to be important in COPD such as IL-8 and TNF-a. Finally, oxidative stress may contribute to reversible airway narrowing. H2O2 constricts airway smooth muscle in vitro and isoprostane F2a-III is a potent constrictor of human airways73.
Pathological changes characteristic of COPD are found in the central airways, peripheral airways, lung parenchyma, and pulmonary vasculature74. The various lesions are a result of chronic inflammation in the lung, which in turn is initiated by the inhalation of noxious particles and gases such as those present in cigarette smoke. The lung has natural defense mechanisms and a considerable capacity to repair itself, but the working of these mechanisms may be affected by genetic traits (e.g., alpha-1 antitrypsin deficiency) or exposure to other environmental risk factors (e.g., infection, atmospheric pollution)75, as well as by the chronic nature of the inflammation and repeated nature of the injury.
The central airways include the trachea, bronchi, and bronchioles greater than 2-4 mm in internal diameter. In patients with chronic bronchitis, an inflammatory exudate of fluid and cells infiltrates the epithelium lining the central airways and associated glands and ducts2,42. The predominant cells in this inflammatory exudate are macrophages and CD8+T lymphocytes2,76. Chronic inflammation in the central airways is also associated with an increase in the number (metaplasia) of epithelial goblet and squamous cells; dysfunction, damage, and/or loss of cilia; enlarged submucosal mucus-secreting glands77; an increase in the amount of smooth muscle and connective tissue in the airway wall78; degeneration of the airway cartilage79, 80; and mucus hypersecretion. The mechanisms of mucus gland hypertrophy and goblet cell metaplasia have not yet been identified, but animal studies81,82 show that irritants including cigarette smoke83 can produce these changes. The various pathological changes in the central airways are responsible for the symptoms of chronic cough and sputum production, which identify people at risk for COPD and may continue to be present throughout the course of the disease. Thus, these pathological changes may be present either on their own or in combination with the changes in the peripheral airways and lung parenchyma described below.
Figure 4.7 Pathological Changes of the Central Airways in COPD

A. Shows a central bronchus from the lung of a cigarette smoker with normal lung function. Only small amounts of muscle are present and the epithelial glands are small. This contrasts sharply with a diseased bronchus.
B. Where the muscle appears as a thick bundle and the glands are enlarged.
C. Shows these enlarged glands at a higher magnification. There is evidence of a chronic inflammatory process involving polymorphonuclear and mononuclear cells, including plasma cells.
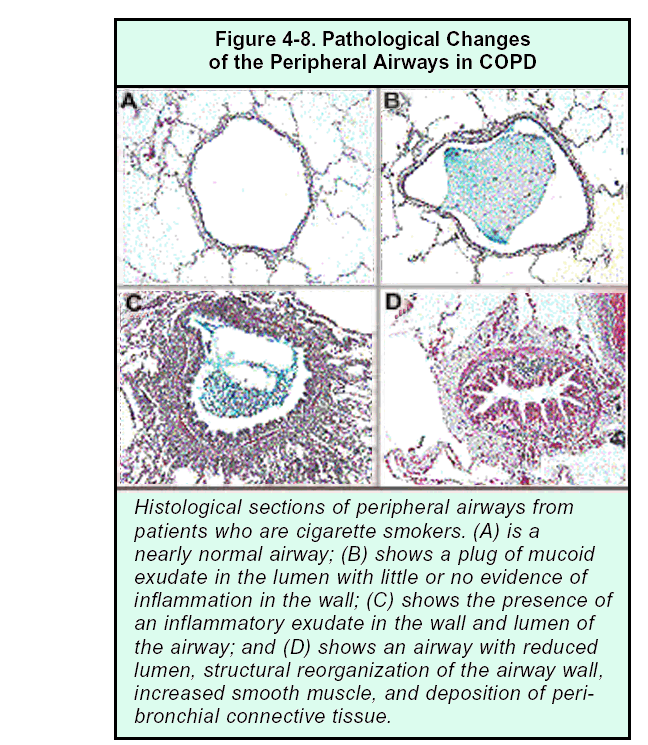
The peripheral airways include small bronchi and bronchioles that have an internal diameter of less than 2 mm (Figure 4-8). The early decline in lung function in COPD is correlated with inflammatory changes in the peripheral airways, similar to those that occur in the central airways: exudate of fluid and cells in the airway wall and lumen, goblet and squamous cell metaplasia of the epithelium43, edema of the airway mucosa due to inflammation, and excess mucus in the airways due to goblet cell metaplasia.
However, the most characteristic change in the peripheral airways of patients with COPD is airway narrowing. Inflammation initiated by cigarette smoking45 and other risk factors75 leads to repeated cycles of injury and repair of the walls of the peripheral airways. Injury is caused either directly by inhaled toxic particles and gases such as those found in cigarette smoke, or indirectly by the action of inflammatory mediators; this injury then initiates repair processes. Although airway repair is only partly understood, it seems likely that disordered repair processes can lead to tissue remodeling with altered structure and function. Cigarette smoke may impair lung repair mechanisms, thereby further contributing to altered lung structure84-86. Even normal lung repair mechanisms can lead to airway remodeling because tissue repair in the airways, as elsewhere in the body, may involve scar tissue formation. In any case, this injury-and-repair process results in a structural remodeling of the airway wall, with increasing collagen content and scar tissue formation, that narrows the lumen and produces fixed airways obstruction87.
The peripheral airways become
the major site of airways obstruction in COPD, and direct measurements of
peripheral airways resistance88 show that the structural changes in
the airway wall are the most important cause of the increase in peripheral
airways resistance in COPD. Inflammatory changes such as airway edema and mucus
hypersecretion also contribute to airway narrowing in COPD. So does loss of
elastic recoil, but fibrosis of the small airways plays the largest role.
Fibrosis in the peripheral airways, as elsewhere in the body, is characterized by the accumulation of mesenchymal cells (fibroblasts and myofibroblasts) and extracellular connective tissue matrix. Several cell types including mononuclear phagocytes and epithelial cells may produce mediators that drive this process. The mediators that drive the accumulation of these cells and of the matrix are incompletely defined, but it is likely that several mediators including TGF-ß, ET-1, Insulin-like growth factor-1, fibronectin, platelet-derived growth factor (PDGF), and others are involved89.
Figure 4.7 Pathological Changes of Peripheral Airways in COPD
The lung parenchyma includes the gas exchanging surface of the lung (respiratory bronchioles and alveoli) and the pulmonary capillary system (Figure 4-9). The most common type of parenchymal destruction in COPD patients is the centrilobular form of emphysema which involves dilatation and destruction of the respiratory bronchioles90. These lesions occur more frequently in the upper lung regions in milder cases, but in advanced disease they may appear diffusely throughout the entire lung and also involve destruction of the pulmonary capillary bed. Panacinar emphysema, which extends throughout the acinus, is the characteristic lesion seen in alpha-1 antitrypsin deficiency and involves dilatation and destruction of the alveolar ducts and sacs as well as the respiratory bronchioles. It tends to affect the lower more than upper lung regions. Because this process usually affects all of the acini in the secondary lobule, it is also referred to as panlobular emphysema. The primary mechanism of lung parenchyma destruction, in both smoking-related COPD and alpha-1 antitrypsin deficiency, is thought to be an imbalance of endogenous proteinases and antiproteinases in the lung. Oxidative stress, another consequence of inflammation, may also contribute91 .
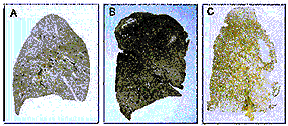 |
Photomicrographs of
paper-mounted whole lung sections prepared from:
A.
normal lung
B.
a lung with mild
centrilobular emphysema, and
C.
a lung with
severe panacinar emphysema.
Note that the centrilobular form affects mainly the upper lung regions whereas the panacinar form is more apparent in the lower lung regions.
Pulmonary vascular changes in COPD (Figure 4-11) are characterized by a thickening of the vessel wall that begins early in the natural history of the disease, when lung function is reasonably well maintained and pulmonary vascular pressures are normal at rest92. Endothelial dysfunction of the pulmonary arteries, which may be caused directly by cigarette smoke products93 or indirectly by inflammatory mediators14, occurs early in COPD94. Since endothelium plays an important role in regulating vascular tone and cell proliferation, it is likely that endothelial dysfunction might initiate the sequence of events that results ultimately in structural changes. Thickening of the intima is the first structural change92, followed by an increase in vascular smooth muscle and the infiltration of the vessel wall by inflammatory cells, including macrophages and CD8+ T lymphocytes14. These structural changes are correlated with an increase in pulmonary vascular pressure that develops first with exercise and then at rest. As COPD worsens, greater amounts of smooth muscle, proteoglycans, and collagen95 further thicken the vessel wall. In advanced disease, the changes in the muscular arteries may be associated with emphysematous destruction of the pulmonary capillary bed.

Pathological changes in COPD lead to corresponding physiological abnormalities that usually become evident first on exercise and later also at rest. Physiological changes characteristic of the disease include mucus hypersecretion, ciliary dysfunction, airflow limitation, pulmonary hyperinflation, gas exchange abnormalities, pulmonary hypertension, and cor pulmonale, and they usually develop in this order over the course of the disease. In turn, various physiological abnormalities contribute to the characteristic symptoms of COPD — chronic cough and sputum production and dyspnea.
Mucus hypersecretion in COPD is caused by the stimulation of the enlarged mucus secreting glands and increased number of goblet cells by inflammatory mediators such as leukotrienes, proteinases, and neuropeptides. Ciliated epithelial cells undergo squamous metaplasia leading to impairment in mucociliary clearance mechanisms. These changes are usually the first physiological abnormalities to develop in COPD, and can be present for many years before any other physiological abnormalities develop.
Expiratory airflow limitation is the hallmark physiological change of COPD. The airflow limitation characteristic of COPD is primarily irreversible, with a small reversible component. Several pathological characteristics contribute to airflow limitation and changes in pulmonary mechanics, as summarized in Figure 4-12. The irreversible component of airflow limitation is primarily due to remodeling42,43,87,88,96,97 — fibrosis and narrowing — of the small airways that produces fixed airways obstruction and a consequent increase in airways resistance. The sites of airflow limitation in COPD are the smaller conducting airways, including bronchi and bronchioles less than 2 mm in internal diameter. In the normal lung, resistance of these smaller airways makes up a small percentage of the total airways resistance88. But in patients with COPD the total lower airways resistance approximately doubles, and most of the increase is due to a large increase in peripheral airways resistance88. Although some have argued that a larger proportion of the total resistance should be attributed to peripheral airways in the normal lung, there is wide agreement that the peripheral airways become the major site of obstruction in COPD.
Figure 4.12 Causes of Airflow Limitation in COPD
|
Irreversible |
· Fibrosis and narrowing of airways · Loss of elastic recoil due to alveolar destruction · Destruction of alveolar support that maintains patency of small airways |
|
Reversible |
· Accumulation of inflammatory cells, mucus, and plasma exudate in bronchi · Smooth muscle contraction in peripheral and central airways · Dynamic hyperinflation during exercise |
Parenchymal destruction (emphysema) plays a smaller role in this irreversible component but contributes to expiratory airflow limitation and the increase in airways resistance in several ways. Destruction of alveolar attachments inhibits the ability of the small airways to maintain patency98. Alveolar destruction is also associated with a loss of elastic recoil of the lung99,100, which decreases the intra-alveolar pressure driving exhalation.
Although both the destruction of alveolar attachments to the outer wall of the peripheral airways and the loss of lung elastic recoil produced by emphysema have been implicated in the pathogenesis of peripheral airways obstruction98,100, direct measurements of peripheral airways resistance88 show that the structural changes in the airway wall are the most important cause of the increase in peripheral airways resistance in COPD.
Airway smooth muscle contraction, ongoing airway inflammation, and intraluminal accumulation of mucus and plasma exudate may be responsible for the small part of airflow limitation that is reversible with treatment. Inflammation and accumulation of mucus and exudate may be particularly important during exacerbations101.
Airflow limitation in COPD is best measured through spirometry, which is key to the diagnosis and management of the disease. The essential spirometric measurements for diagnosis and monitoring of COPD patients are the forced expiratory volume in one second (FEV1) and forced vital capacity (FVC). As COPD progresses, with increased thickness of the airway wall, loss of alveolar attachments, and loss of lung elastic recoil, FEV1 and FVC decrease. A decrease in the ratio of FEV1 to FVC is often the first sign of developing airflow limitation. FEV1 declines naturally with age, but the rate of decline in COPD patients is generally greater than that in normal subjects.
With increasing severity of airflow limitation, expiration becomes flow-limited during tidal breathing. Initially, this occurs only during exercise, but later it is also seen at rest. In parallel with this, functional residual capacity (FRC) increases due to the combination of the decrease in the elastic properties of the lungs, premature airway closure, and a variable dynamic element reflecting the breathing pattern adopted to cope with impaired lung mechanics. As airflow limitation develops, the rate of lung emptying is slowed and the interval between inspiratory efforts does not allow expiration to the relaxation volume of the respiratory system; this leads to dynamic pulmonary hyperinflation. The increase in FRC can impair inspiratory muscle function and coordination, although the contractility of the diaphragm, when normalized for lung volume, seems to be preserved. These changes occur as the disease advances but are almost always seen first during exercise, when the greater metabolic stimulus to ventilation stresses the ability of the ventilatory pump to maintain gas exchange.
In advanced COPD, peripheral airways obstruction, parenchymal destruction, and pulmonary vascular abnormalities reduce the lung’s capacity for gas exchange, producing hypoxemia and, later on, hypercapnia. The correlation between routine lung function tests and arterial blood gases is poor, but significant hypoxemia or hypercapnia is rare when FEV1 is greater than 1.00 L102. Hypoxemia is initially only present during exercise, but as the disease continues to progress it is also present at rest.
Inequality in the ventilation/perfusion ratio (VA/Q) is the major mechanism behind hypoxemia in COPD, regardless of the stage of the disease103. In the peripheral airways, injury of the airway wall is associated with VA/Q mismatching, as indicated by a significant correlation between bronchiolar inflammation and the distribution of ventilation. In the parenchyma, destruction of the lung surface area by emphysema reduces diffusing capacity and interferes with gas exchange104. High VA/Q units probably represent emphysematous regions with alveolar destruction and loss of pulmonary vasculature. The severity of pulmonary emphysema appears to be related to the overall inefficiency of the lung as a gas exchanger. This is reflected by the good correlation between the diffusing capacity of carbon monoxide per liter of alveolar volume (DLco/VA) and the severity of macroscopic emphysema. Reduced ventilation due to loss of elastic recoil in the emphysematous lung, together with the loss of the capillary bed and the generalized inhomogeneity of ventilation due to the patchy nature of these changes, leads to areas of VA/Q mismatching that result in arterial hypoxemia.
The relationship between pulmonary vascular abnormalities and VA/Q relationships has been investigated in patients with mild COPD. The more severe the vessel wall damage is, the less the reversal of hypoxic vasoconstriction by oxygen105. This suggests that pathology in the pulmonary artery wall, particularly when it affects the intimal layer, may play a key role in determining the loss of vascular response to hypoxia that contributes to VA/Q mismatching. Chronic hypercapnia usually reflects inspiratory muscle dysfunction and alveolar hypoventilation.
Pulmonary hypertension develops late in the course of COPD (Stage III: Severe COPD), usually after the development of severe hypoxemia (PaO2 < 8.0 kPa or 60 mm Hg) and often hypercapnia as well. It is the major cardiovascular complication of COPD and is associated with the development of cor pulmonale and with a poor prognosis106. However, even in patients with severe disease, pulmonary arterial pressure is usually only modestly elevated at rest, though it may rise markedly with exercise. Pulmonary hypertension in COPD is believed to progress rather slowly even if left untreated. Further studies are required to firmly establish the natural history of pulmonary hypertension in COPD.
Factors that are known to contribute to the development of pulmonary hypertension in patients with COPD include vasoconstriction; remodeling of pulmonary arteries, which thickens the vessel walls and reduces the lumen; and destruction of the pulmonary capillary bed by emphysema, which further increases the pressure required to perfuse the pulmonary vascular bed. Vasoconstriction may itself have several causes, including hypoxia, which causes pulmonary vascular smooth muscle to contract; impaired mechanisms of endothelium-dependent vasodilation, such as reduced NO synthesis or release; and abnormal secretion of vasoconstrictor peptides (such as ET-1, which is produced by inflammatory cells). In advanced COPD, hypoxia plays the primary role in producing pulmonary hypertension, both by causing vasoconstriction of the pulmonary arteries and by promoting remodeling of the vessel wall (either by inducing the release of growth factors107 or as a consequence of the mechanical stress that results from hypoxic vasoconstriction).
Pulmonary hypertension is associated with the development of cor pulmonale, defined as “hypertrophy of the right ventricle resulting from diseases affecting the function and/or structure of the lungs, except when these pulmonary alterations are the result of diseases that primarily affect the left side of the heart, as in congenital heart disease.” This is a pathological definition and the clinical diagnosis and assessment of right ventricular hypertrophy is difficult in life.
The prevalence and natural history of cor pulmonale in COPD are not yet clear. Pulmonary hypertension and reduction of the vascular bed due to emphysema can lead to right ventricular hypertrophy and right heart failure, but right ventricular function appears to be maintained in some patients despite the presence of pulmonary hypertension108. Right heart failure is associated with venous stasis and thrombosis that may result in pulmonary embolism and further compromise the pulmonary circulation.
COPD is associated with systemic (i.e., extrapulmonary) effects, such as systemic inflammation and skeletal muscle dysfunction. Evidence of systemic inflammation includes the presence of systemic oxidative stress109, abnormal concentrations of circulating cytokines110, and activation of inflammatory cells111,112. Evidence of skeletal muscle dysfunction includes the progressive loss of skeletal muscle mass and the presence of several bioenergetic abnormalities113. These systemic effects have important clinical consequences, as they contribute to the limitation of patients’ exercise capacity and thus the decline of health status in COPD. The presence of these systemic effects appears to worsen a patient’s prognosis114.
Chronic cough and sputum production, sometimes labeled as chronic bronchitis, are a result of airway inflammation, which leads to mucus hypersecretion and dysfunction of the normal ciliary clearance mechanisms. Sputum is produced in COPD as a result of the inflammatory response, and contains plasma proteins exuded from the microvessels of the bronchial circulation, inflammatory cells, and small amounts of mucus from epithelial goblet cells. The volume of sputum produced overpowers clearance mechanisms, resulting in cough and expectoration. Some pathological abnormalities, such as inflammation of the submucosal glands and hyperplasia of goblet cells, may contribute to chronic sputum production, although these pathological abnormalities are not present in all patients with this symptom.
Dyspnea, an abnormal awareness of the act of breathing, usually reflects an imbalance between the neural drive to the respiratory muscles and the effectiveness of the resulting ventilation. Different individuals use different words to describe the feeling of breathlessness, which is also influenced by other factors such as mood. In COPD patients, dyspnea is mainly the result of impaired lung mechanics (increased airways resistance, decreased elastic recoil). It is only present on vigorous exercise in the early stages of disease but may be present at rest as the mechanical impairment becomes severe.
The progressive course of COPD is complicated by acute exacerbations that have many causes and occur with increasing frequency as the disease progresses.
Distinguishing the pathology of these acute events from that of the underlying disease is difficult because patients experiencing an exacerbation are usually too ill to study. The limited evidence available suggests that mild COPD exacerbations are associated with increases of both neutrophils and eosinophils in sputum and biopsies, while severe COPD exacerbations are associated with an increase in sputum neutrophils and eosinophils18,19. At least in sputum, the changes in inflammatory cells during exacerbations of COPD are the same as those observed during exacerbations of asthma115-119. So far no study has been conducted examining the pathological abnormalities associated with fatal exacerbations of COPD, which can be considered the extreme end of the spectrum of severity.
Expiratory airflow is almost unchanged during mild exacerbations18, and only slightly reduced during severe exacerbations120,121. Although the pathophysiology of acute exacerbations is not fully understood, the primary physiological change in severe acute exacerbations is a further worsening of gas exchange, primarily produced by increased VA/Q inequality. As VA /Q relationships worsen, increased work of the respiratory muscles results in greater oxygen consumption, decreased mixed venous oxygen tension, and further amplification of gas exchange abnormalities120. Worsening of VA/Q relationships has several causes in acute exacerbations. Airway inflammation and edema, mucus hypersecretion, and bronchoconstriction may contribute to changes in the distribution of ventilation, while hypoxic constriction of pulmonary arterioles may modify the distribution of perfusion. Additional contributors to worsening gas exchange in acute exacerbations include abnormal patterns of breathing and fatigue of the respiratory muscles. These can cause further deterioration in blood gases and worsening of respiratory acidosis, leading to severe respiratory failure and death120-123. Alveolar hypoventilation also contributes to hypoxemia, hypercapnia, and respiratory acidosis. In turn, hypoxemia and respiratory acidosis promote pulmonary vasoconstriction, which increases pulmonary artery pressures and imposes an added load on the right ventricle.
1. Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med 1995; 152:1666-72.
2. O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med 1997; 155:852-7.
3. Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998; 158:1277-85.
4. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153:530-4.
5. Thompson AB, Daughton D, Robbins RA, Ghafouri MA, Oehlerking M, Rennard SI. Intraluminal airway inflammation in chronic bronchitis. Characterization and correlation with clinical parameters. Am Rev Respir Dis 1989; 140:1527-37.
6. Lacoste JY, Bousquet J, Chanez P, Van Vyve T, Simony-Lafontaine J, Lequeu N, et al. Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary disease. J Allergy Clin Immunol 1993; 92:537-48.
7. Pesci A, Balbi B, Majori M, Cacciani G, Bertacco S, Alciato P, et al. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J 1998; 12:380-6.
8. Pesci A, Majori M, Cuomo A, Borciani N, Bertacco S, Cacciani G, et al. Neutrophils infiltrating bronchial epithelium in chronic obstructive pulmonary disease. Respir Med 1998; 92:863-70.
9. Peleman RA, Rytila PH, Kips JC, Joos GF, Pauwels RA. The cellular composition of induced sputum in chronic obstructive pulmonary disease. Eur Respir J 1999; 13:839-43.
10. Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157:822-6.
11. Roth MD, Arora A, Barsky SH, Kleerup EC, Simmons M, Tashkin DP. Airway inflammation in young marijuana and tobacco smokers. Am J Respir Crit Care Med 1998; 157:928-37.
12. Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med 1997; 155:449-53.
13. Balbi B, Bason C, Balleari E, Fiasella F, Pesci A, Ghio R, et al. Increased bronchoalveolar granulocytes and granulocyte/macrophage colony-stimulating factor during exacerbations of chronic bronchitis. Eur Respir J 1997; 10:846-50.
14. Peinado VI, Barbera JA, Abate P, Ramirez J, Roca J, Santos S, et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159:1605-11.
15. Liu AN, Mohammed AZ, Rice WR, Fiedeldey DT, Liebermann JS, Whitsett JA, et al. Perforin-independent CD8(+) T-cell-mediated cytotoxicity of alveolar epithelial cells is preferentially mediated by tumor necrosis factor-alpha: relative insensitivity to Fas ligand. Am J Respir Cell Mol Biol 1999; 20:849-58.
16. Mueller R, Chanez P, Campbell AM, Bousquet J, Heusser C, Bullock GR. Different cytokine patterns in bronchial biopsies in asthma and chronic bronchitis. Respir Med 1996; 90:79-85.
17. Liu H, Lazarus SC, Caughey GH, Fahy JV. Neutrophil elastase and elastase-rich cystic fibrosis sputum degranulate human eosinophils in vitro. Am J Physiol 1999; 276:L28-34.
18. Saetta M, Di Stefano A, Maestrelli P, Turato G, Ruggieri MP, Roggeri A, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med 1994; 150:1646-52.
19. Saetta M, Di Stefano A, Maestrelli P, Turato G, Mapp CE, Pieno M, et al. Airway eosinophilia and expression of interleukin-5 protein in asthma and in exacerbations of chronic bronchitis. Clin Exp Allergy 1996; 26:766-74.
20. Mills PR, Davies RJ, Devalia JL. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med 1999; 160:S38-43.
21. Di Stefano A, Maestrelli P, Roggeri A, Turato G, Calabro S, Potena A, et al. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med 1994; 149:803-10.
22. McElvaney NG, Crystal RG. Proteases and lung injury. In: Crystal RG, West JB, eds. The lung: scientific foundations. Philadelphia: Lippincott-Raven; 1997. p. 2205-18.
23. Shapiro SD. Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr Opin Cell Biol 1998; 10:602-8.
24. Warren JS, Ward PA. Consequences of oxidant injury. In: Crystal RG, West JB, eds. The lung: scientific foundations. Philadelphia: Lippincott-Raven; 1997. p. 2279-88.
25. Ganz T, Lehrer RI. Defensins. Curr Opin Immunol 1994; 6:584-9.
26. Hill AT, Bayley D, Stockley RA. The interrelationship of sputum inflammatory markers in patients with chronic bronchitis. Am J Respir Crit Care Med 1999; 160:893-8.
27. Yamamoto C, Yoneda T, Yoshikawa M, Fu A, Tokuyama T, Tsukaguchi K, et al. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest 1997; 112:505-10.
28. Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest 1980; 66:859-62.
29. Heise CE, O’Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, et al. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem 2000; 275:30531-6.
30. de Godoy I, Donahoe M, Calhoun WJ, Mancino J, Rogers RM. Elevated TNF-alpha production by peripheral blood monocytes of weight-losing COPD patients. Am J Respir Crit Care Med 1996; 153:633-7.
31. Capelli A, Di Stefano A, Gnemmi I, Balbo P, Cerutti CG, Balbi B, et al. Increased MCP-1 and MIP-1beta in bronchoalveolar lavage fluid of chronic bronchitics. Eur Respir J 1999; 14:160-5.
32. Hoshi H, Ohno I, Honma M, Tanno Y, Yamauchi K, Tamura G, et al. IL-5, IL-8 and GM-CSF immunostaining of sputum cells in bronchial asthma and chronic bronchitis. Clin Exp Allergy 1995; 25:720-8.
33. Vignola AM, Chanez P, Chiappara G, Merendino A, Pace E, Rizzo A, et al. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med 1997; 156:591-9.
34. de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PS, et al. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 158:1951-7.
35. Chalmers GW, Macleod KJ, Sriram S, Thomson LJ, McSharry C, Stack BH, et al. Sputum endothelin-1 is increased in cystic fibrosis and chronic obstructive pulmonary disease. Eur Respir J 1999; 13:1288-92.
36. Fujii T, Otsuka T, Tanaka S, Kanazawa H, Hirata K, Kohno M, et al. Plasma endothelin-1 level in chronic obstructive pulmonary disease: relationship with natriuretic peptide. Respiration 1999; 66:212-9.
37. Tomaki M, Ichinose M, Miura M, Hirayama Y, Yamauchi H, Nakajima N, et al. Elevated substance P content in induced sputum from patients with asthma and patients with chronic bronchitis. Am J Respir Crit Care Med 1995; 151:613-7.
38. Lucchini RE, Facchini F, Turato G, Saetta M, Caramori G, Ciaccia A, et al. Increased VIP-positive nerve fibers in the mucous glands of subjects with chronic bronchitis. Am J Respir Crit Care Med 1997; 156:1963-8.
39. Chanez P, Springall D, Vignola AM, Moradoghi-Hattvani A, Polak JM, Godard P, et al. Bronchial mucosal immunoreactivity of sensory neuropeptides in severe airway diseases. Am J Respir Crit Care Med 1998; 158:985-90.
40. Metcalf JP, Thompson AB, Gossman GL, Nelson KJ, Koyama S, Rennard SI, et al. Gcglobulin functions as a cochemotaxin in the lower respiratory tract. A potential mechanism for lung neutrophil recruitment in cigarette smokers. Am Rev Respir Dis 1991; 143:844-9.
41. Wright JL, Lawson LM, Pare PD, Wiggs BJ, Kennedy S, Hogg JC. Morphology of peripheral airways in current smokers and ex-smokers. Am Rev Respir Dis 1983; 127:474-7.
42. Mullen JB, Wright JL, Wiggs BR, Pare PD, Hogg JC. Reassessment of inflammation of airways in chronic bronchitis. BMJ (Clin Res Ed) 1985; 291:1235-9.
43. Cosio M, Ghezzo H, Hogg JC, Corbin R, Loveland M, Dosman J, et al. The relations between structural changes in small airways and pulmonary-function tests. N Engl J Med 1978; 298:1277-81.
44. McLean KA. Pathogenesis of pulmonary emphysema. Am J Med 1958; 25:62-74.
45. Niewoehner DE, Kleinerman J, Rice DB. Pathologic changes in the peripheral airways of young cigarette smokers. N Engl J Med 1974; 291:755-8.
46. Auerbach O, Garfinkel L, Hammond EC. Relation of smoking and age to findings in lung parenchyma: a microscopic study. Chest 1974; 65:29-35.
47. Auerbach O, Hammond EC, Garfinkel L, Benante C. Relation of smoking and age to emphysema. Whole-lung section study. N Engl J Med 1972; 286:853-7.
48. Petty TL, Silvers GW, Stanford RE, Baird MD, Mitchell RS. Small airway pathology is related to increased closing capacity and abnormal slope of phase III in excised human lungs. Am Rev Respir Dis 1980; 121:449-56.
49. Ollerenshaw SL, Woolcock AJ. Characteristics of the inflammation in biopsies from large airways of subjects with asthma and subjects with chronic airflow limitation. Am Rev Respir Dis 1992; 145:922-7.
50. Gadek JE, Fells GA, Crystal RG. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science 1979; 206:1315-6.
51. Stone PJ, Calore JD, McGowan SE, Bernardo J, Snider GL, Franzblau C. Functional alpha 1-protease inhibitor in the lower respiratory tract of cigarette smokers is not decreased. Science 1983; 221:1187-9.
52. Hunninghake GW, Crystal RG. Cigarette smoking and lung destruction. Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis 1983; 128:833-8.
53. Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med 1997; 155:1770-6.
54. Masubuchi T, Koyama S, Sato E, Takamizawa A, Kubo K, Sekiguchi M, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol 1998; 153:1903-12.
55. Speizer FE, Tager IB. Epidemiology of chronic mucus hypersecretion and obstructive airways disease. Epidemiol Rev 1979; 1:124-42.
56. Turato G, Di Stefano A, Maestrelli P, Mapp CE, Ruggieri MP, Roggeri A, et al. Effect of smoking cessation on airway inflammation in chronic bronchitis. Am J Respir Crit Care Med 1995; 152:1262-7.
57. Li XY, Brown D, Smith S, MacNee W, Donaldson K. Short-term inflammatory responses following intratracheal instillation of fine and ultrafine carbon black in rats Inhal Toxicol 1999; 11:709-31.
58. Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol1999; 155:245-52.
59. Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med 1999; 159:702-9.
60. Von Essen SG, O’Neill DP, McGranaghan S, Olenchock SA, Rennard SI. Neutrophilic respiratory tract inflammation and peripheral blood neutrophilia after grain sorghum dust extract challenge. Chest 1995; 108:1425-33.
61. Von Essen SG, Robbins RA, Thompson AB, Ertl RF, Linder J, Rennard SI. Mechanisms of neutrophil recruitment to the lung by grain dust exposure [published erratum appears in Am Rev Respir Dis 1989; 139:1065]. Am Rev Respir Dis 1988; 138:921-7.
62. Laurell CB, Eriksson S. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132-40.
63. Carp H, Miller F, Hoidal JR, Janoff A. Potential mechanism of emphysema: alpha 1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci U S A 1982; 79:2041-5.
64. Cohen AB, James HL. Reduction of the elastase inhibitory capacity of alpha 1-antitrypsin by peroxides in cigarette smoke: an analysis of brands and filters. Am Rev Respir Dis 1982; 126:25-30.
65. Shapiro SD. Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease. Am J Respir Crit Care Med 1994; 150:S160-4.
66. Sommerhoff CP, Nadel JA, Basbaum CB, Caughey GH. Neutrophil elastase and cathepsin G stimulate secretion from cultured bovine airway gland serous cells. J Clin Invest 1990; 85:682-9.
67. Witko-Sarsat V, Halbwachs-Mecarelli L, Schuster A, Nusbaum P, Ueki I, Canteloup S, et al. Proteinase 3, a potent secretagogue in airways, is present in cystic fibrosis sputum. Am J Respir Cell Mol Biol 1999; 20:729-36.
68. Christensen TG, Korthy AL, Snider GL, Hayes JA. Irreversible bronchial goblet cell metaplasia in hamsters with elastase-induced panacinar emphysema. J Clin Invest 1977; 59:397-404.
69. Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL, et al. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1996; 154:813-6.
70. Maziak W, Loukides S, Culpitt S, Sullivan P, Kharitonov SA, Barnes PJ. Exhaled nitric oxide in chronic obstructive pulmonary disease.Am J Respir Crit Care Med 1998; 157:998-1002.
71. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, et al. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med 2000; 162:1175-7.
72. Pratico D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald GA. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stress. Am J Respir Crit Care Med 1998; 158:1709-14.
73. Kawikova I, Barnes PJ, Takahashi T, Tadjkarimi S, Yacoub MH, Belvisi MG. 8-Epi-PGF2 alpha, a novel noncyclooxygenase-derived prostaglandin, constricts airways in vitro. Am J Respir Crit Care Med 1996; 153:590-6.
74. Ciba Guest Symposium Report. Terminiology, definitions and classifications of chronic pulmonary emphysema and related conditions. Thorax 1959; 14:286-99.
75. Pride NB, Burrows B. Development of impaired lung function: natural history and risk factors. In: Calverley PM, Pride NB, eds. Chronic obstructive lung disease. London and Glasgow: Chapman and Hall Medical; 1995. p. 69-71.
76. Saetta M, Di Stefano A, Maestrelli P, Ferraresso A, Drigo R, Potena A, et al. Activated T-lymphocytes and macrophages in bronchial mucosa of subjects with chronic bronchitis. Am Rev Respir Dis 1993; 147:301-6.
77. Reid L. Measurement of the bronchial mucous gland layer: a diagnostic yardstick in chronic bronchitis. Thorax 1960; 15:132-41.
78. Jamal K, Cooney TP, Fleetham JA, Thurlbeck WM. Chronic bronchitis. Correlation of morphologic findings to sputum production and flow rates. Am Rev Respir Dis 1984; 129:719-22.
79. Haraguchi M, Shimura S, Shirato K. Morphometric analysis of bronchial cartilage in chronic obstructive pulmonary disease and bronchial asthma.Am J Respir Crit Care Med 1999; 159:1005-13.
80. Thurlbeck WM, Pun R, Toth J, Frazer RG. Bronchial cartilage in chronic obstructive lung disease. Am Rev Respir Dis 1974; 109:73-80.
81. Snider GL. Parker B. Francis Lecture. Animal models of chronic airways injury. Chest 1992; 101:74-9S.
82. Snider GL, Faling LJ, Rennard SI. Chronic bronchitis and emphysema. In: Murray JF, Nadel JA, eds. Textbook of respiratory medicine. Philadelphia: WB Saunders; 2000. p. 1187-246.
83. Rogers DF, Jeffery PK. Inhibition by oral N-acetylcysteine of cigarette smoke-induced “bronchitis” in the rat. Exp Lung Res 1986; 10:267-83.
84. Nakamura Y, Romberger DJ, Tate L, Ertl RF, Kawamoto M, Adachi Y, et al. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med 1995; 151:1497-503.
85. Osman M, Cantor JO, Roffman S, Keller S, Turino GM, Mandl I. Cigarette smoke impairs elastin resynthesis in lungs of hamsters with elastase-induced emphysema. Am Rev Respir Dis 1985; 132:640-3.
86. Laurent P, Janoff A, Kagan HM. Cigarette smoke blocks cross-linking of elastin in vitro. Chest 1983; 83:63-5S.
87. Matsuba K, Thurlbeck WM. The number and dimensions of small airways in emphysematous lungs. Am J Pathol 1972; 67:265-75.
88. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 1968; 278:1355-60.
89. Rennard SI. Inflammation and repair processes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 160:S12-6.
90. Leopold JG, Goeff J. Centrilobular form of hypertrophic emphysema and its relation to chronic bronchitis. Thorax 1957; 12:219-35.
91. Repine JE, Bast A, Lankhorst I. Oxidative stress in chronic obstructive pulmonary disease. Oxidative Stress Study Group. Am J Respir Crit Care Med 1997; 156:341-57.
92. Wright JL, Lawson L, Pare PD, Hooper RO, Peretz DI, Nelems JM, et al. The structure and function of the pulmonary vasculature in mild chronic obstructive pulmonary disease. The effect of oxygen and exercise. Am Rev Respir Dis 1983; 128:702-7.
93. Sekhon HS, Wright JL, Churg A. Cigarette smoke causes rapid cell proliferation in small airways and associated pulmonary arteries. Am J Physiol 1994; 267:L557-63.
94. Peinado VI, Barbera JA, Ramirez J, Gomez FP, Roca J, Jover L, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol 1998; 274:L908-13.
95. Riley DJ, Thakker-Varia S, Poiani GJ, Tozzi CA. Vascular remodeling. In: Crystal RG, West JB, Barnes PJ, Weibel ER, eds. The lung: scientific foundations. Philadelphia: Lippincott-Raven; 1977. p. 1589-97.
96. Kuwano K, Bosken CH, Pare PD, Bai TR, Wiggs BR, Hogg JC. Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis 1993; 148:1220-5.
97. Matsuba K, Wright JL, Wiggs BR, Pare PD, Hogg JC. The changes in airways structure associated with reduced forced expiratory volume in one second. Eur Respir J 1989; 2:834-9.
98. Dayman H. Mechanics of airflow in health and emphysema. J Clin Invest 1951; 30:1175-90.
99. Butler J, Caro C, Alkaler R, Dubois AB. Physiological factors affecting airway resistance in normal subjects and in patients with obstructive airways disease. J Clin Invest 1960; 39:584-91.
100. Mead J, Turner JM, Macklem PT, Little JB. Significance of the relationship between lung recoil and maximum expiratory flow. J Appl Physiol 1967; 22:95-108.
101. Burnett D, Stockley RA. Serum and sputum alpha 2 macroglobulin in patients with chronic obstructive airways disease. Thorax 1981; 36:512-6.
102. Lane DJ, Howell JB, Giblin B. Relation between airways obstruction and CO2 tension in chronic obstructive airways disease. BMJ 1968; 3:707-9.
103. Rodriguez-Roisin R, MacNee W. Pathophysiology of chronic obstructive pulmonary disease. In: Postma DS, Siafakas NM, eds. Management of chronic obstructive pulmonary disease. European Respiratory Monograph 1998; 3:107-26.
104. McLean A, Warren PM, Gillooly M, MacNee W, Lamb D. Microscopic and macroscopic measurements of emphysema: relation to carbon monoxide gas transfer. Thorax 1992; 47:144-9.
105. Barbera JA, Riverola A, Roca J, Ramirez J, Wagner PD, Ros D, et al. Pulmonary vascular abnormalities and ventilation-perfusion relationships in mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1994; 149:423-9.
106. MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part two. Am J Respir Crit Care Med 1994; 150:1158-68.
107. Knighton DR, Hunt TK, Scheuenstuhl H, Halliday BJ, Werb Z, Banda MJ. Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science 1983; 221:1283-5.
108. Biernacki W, Flenley DC, Muir AL, MacNee W. Pulmonary hypertension and right ventricular function in patients with COPD. Chest 1988; 94:1169-75.
109. Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996; 154:1055-60.
110. Schols AM, Buurman WA, Staal van den Brekel AJ, Dentener MA, Wouters EF. Evidence for a relation between metabolic derangements and increased levels of inflammatory mediators in a subgroup of patients with chronic obstructive pulmonary disease. Thorax 1996; 51:819-24.
111. .Sauleda J, Garcia-Palmer FJ, Gonzalez G, Palou A, Agusti AG. The activity of cytochrome oxidase is increased in circulating lymphocytes of patients with chronic obstructive pulmonary disease, asthma, and chronic arthritis. Am J Respir Crit Care Med 2000; 161:32-5.
112. Sauleda J, Garcia-Palmer F, Wiesner RJ, Tarraga S, Harting I, Tomas P, et al. Cytochrome oxidase activity and mitochondrial gene expression in skeletal muscle of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157:1413-7.
113. American Thoracic Society and European Respiratory Society. Skeletal muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159:S1-40.
114. Schols AM, Slangen J, Volovics L, Wouters EF. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157:1791-7.
115. Pizzichini MM, Pizzichini E, Efthimiadis A, Clelland L, Mahony JB, Dolovich J, et al. Markers of inflammation in induced sputum in acute bronchitis caused by Chlamydia pneumoniae. Thorax 1997; 52:929-31; discussion 6-7.
116. Pizzichini E, Pizzichini MM, Gibson P, Parameswaran K, Gleich GJ, Berman L, et al. Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitis. Am J Respir Crit Care Med 1998; 158:1511-7.
117. Maestrelli P, Saetta M, Di Stefano A, Calcagni PG, Turato G, Ruggieri MP, et al. Comparison of leukocyte counts in sputum, bronchial biopsies, and bronchoalveolar lavage. Am J Respir Crit Care Med 1995; 152:1926-31.
118. Turner MO, Hussack P, Sears MR, Dolovich J, Hargreave FE. Exacerbations of asthma without sputum eosinophilia. Thorax 1995; 50:1057-61.
119. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol 1995; 95:843-52.
120. Barbera JA, Roca J, Ferrer A, Felez MA, Diaz O, Roger N, et al. Mechanisms of worsening gas exchange during acute exacerbations of chronic obstructive pulmonary disease. Eur Respir J 1997; 10:1285-91.
121. Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 161:1608-13.
122. Schmidt GA, Hall JB. Acute or chronic respiratory failure. Assessment and management of patients with COPD in the emergency setting. JAMA 1989; 261:3444-53.
123. Rodriguez-Roisin R. Pulmonary gas exchange in acute respiratory failure. Eur J Anaesthesiol 1994; 11:5-13.
| Course |