| Course |
· Risk factors for COPD include both host factors and environmental exposures, and the disease usually arises from an interaction between these two types of factors.
· The host factor that is best documented is a rare hereditary deficiency of alpha-1 antitrypsin. Other genes involved in the pathogenesis of COPD have not yet been identified.
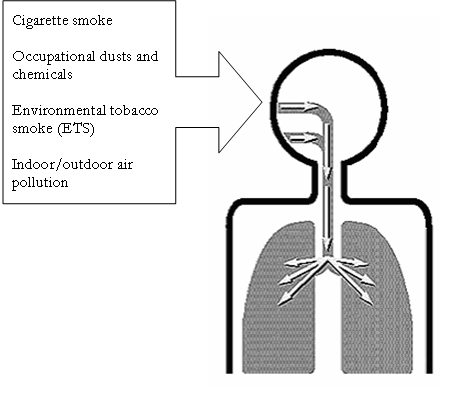
· The major environmental factors are tobacco smoke, occupational dusts and chemicals (vapors, irritants, fumes), and indoor/outdoor air pollution.
The identification of risk factors is an important step toward developing strategies for prevention and treatment of any disease. Identification of cigarette smoking as an important risk factor for COPD has led to the incorporation of smoking cessation programs as a key element of COPD prevention, as well as an important intervention for patients who already have the disease. However, although smoking is the best-studied COPD risk factor, it is not the only one. Further studies of other risk factors could lead to similar powerful interventions.
Much of the evidence concerning risk factors for COPD comes from cross-sectional epidemiological studies that identify associations rather than cause-and-effect relationships. Although several longitudinal studies (which are capable of revealing causal relationships) of COPD have followed groups and populations for up to 20 years, none of them has monitored the progression of the disease through its entire course. Thus, current understanding of risk factors for COPD is in many respects incomplete.
The division into “Host Factors” and “Exposures” reflects the current understanding of COPD as resulting from an interaction between the two types of factors. Thus, of two people with the same smoking history, only one may develop COPD due to differences in genetic predisposition to the disease, or in how long they live. Risk factors for COPD may also be related in more complex ways. For example, gender may influence whether a person takes up smoking or experiences certain occupational or environmental exposures; socioeconomic status may be linked to a child’s birth weight; longer life expectancy will allow greater lifetime exposure to risk factors; etc. Understanding the relationships and interactions among risk factors is a crucial area of ongoing investigation.
The best-documented host factor is a severe hereditary deficiency of alpha-1 antitrypsin. The major environmental factors are tobacco smoke, occupational dusts and chemicals (vapors, irritants, fumes), and indoor and outdoor air pollution. However, it is very difficult to demonstrate that a given risk factor is sufficient to cause the disease.
Data are not available to determine whether the increasing prevalence of respiratory symptoms and the accelerated rate of lung function decline that occur with age reflect the cumulative exposure to respiratory particles, irritants, fumes, vapors, etc., or host-related phenomena such as the loss of elastic recoil of lung tissue and stiffening of the chest wall. The field of normal lung aging has been only minimally explored and more work is required.
The role of gender as a risk factor for COPD remains unclear. In the past, most studies showed that COPD prevalence and mortality were greater among men than women1-4. More recent studies5,6 from developed countries show that the prevalence of the disease is almost equal in men and women, which probably reflects changing patterns of tobacco smoking. Some studies have in fact suggested that women are more susceptible to the effects of tobacco smoke than men4,7. This is an important question given the increasing rate of smoking among women in both developed and developing countries.
The role of nutritional status as an independent risk factor for the development of COPD is unclear. Malnutrition and weight loss can reduce respiratory muscle strength and endurance, apparently by reducing both respiratory muscle mass and the strength of the remaining muscle fibers8. The association of starvation and anabolic/catabolic status with the development of emphysema has been shown in experimental studies in animals9.
It is believed that many genetic factors increase (or decrease) a person’s risk of developing COPD. Studies have demonstrated an increased risk of COPD within families with COPD probands. Some of this risk may be due to shared environmental factors, but several studies in diverse populations also suggest a shared genetic risk10,11.
The genetic risk factor that is best documented is a severe hereditary deficiency of alpha-1 antitrypsin12-14, a major circulating inhibitor of serine proteases. This rare hereditary deficiency is a recessive trait most commonly seen in individuals of Northern European origin. Premature and accelerated development of panlobular emphysema and decline in lung function occur in both smokers and nonsmokers with the severe deficiency, although smoking increases the risk appreciably. There is considerable variation between individuals in the extent and severity of the emphysema and the rate of lung function decline. Although alpha-1 antitrypsin deficiency is relevant to only a small part of the world’s population, it illustrates the interaction between host factors and environmental exposures leading to COPD. In this way, it provides a model for how other genetic risk factors are thought to contribute to COPD.
Exploratory studies have revealed a
number of candidate genes that may influence a person’s risk of COPD, including
ABO secretor status15,16, microsomal epoxide hydrolase17,
glutathione S-transferase18, alpha-1 antichymotrypsin19,
the complement component GcG20, cytokine TNF-![]() 21, and
micro-satellite instability22. However, when several studies of a
given trait are available, the results are often inconsistent. Several of these
genes are thought to be involved in inflammation, and therefore are related to
potential pathogenic mechanisms of COPD.
21, and
micro-satellite instability22. However, when several studies of a
given trait are available, the results are often inconsistent. Several of these
genes are thought to be involved in inflammation, and therefore are related to
potential pathogenic mechanisms of COPD.
Asthma and airway hyperresponsiveness, identified as risk factors that contribute to the development of COPD, are complex disorders related to a number of genetic and environmental factors. The relationship between asthma/airway hyperresponsiveness and increased risk of developing COPD was originally described by Orie and colleagues23 and termed the “Dutch hypothesis.” Asthmatics, as a group, experience a slightly accelerated loss of lung function24,25 compared to non-asthmatics, as do smokers with airway hyperresponsiveness compared to normal smokers26. How these trends are related to the development of COPD is unknown, however. Airway hyperresponsiveness may also develop after exposure to tobacco smoke or other environmental insults and thus may be a result of smoking-related airway disease.
Lung growth is related to processes occurring during gestation, birth weight, and exposures during childhood27-31. Reduced maximal attained lung function (as measured by spirometry) may identify individuals who are at increased risk for the development of COPD32.
It may be helpful conceptually to think of a person’s exposures in terms of his or her total burden of inhaled particles. Each type of particle, depending on its size and composition, may contribute a different weight to the risk, and the total risk will depend on the integral of the inhaled exposures. Of the many inhalational exposures that people may encounter over a lifetime, only tobacco smoke2,33-39 and occupational dusts and chemicals (vapors, irritants, and fumes)40,41 are known to cause COPD on their own. Tobacco smoke and occupational exposures also appear to act additively to increase a person’s risk of developing COPD.
Figure 3.2 Total Burden of Inhaled Particles
 |
Cigarette smoking is by far the most important risk factor for COPD and the most important way that tobacco contributes to the risk of COPD. Cigarette smokers have a higher prevalence of respiratory symptoms and lung function abnormalities, a greater annual rate of decline in FEV1, and a greater COPD mortality rate than nonsmokers. These differences between cigarette smokers and nonsmokers increase in direct proportion to the quantity of smoking. Pipe and cigar smokers have greater COPD morbidity and mortality rates than nonsmokers, although their rates are lower than those for cigarette smokers33.
Other types of tobacco smoking popular in various countries are also risk factors for COPD, although their risk relative to cigarette smoking has not been reported.
Age at starting to smoke, total pack-years smoked, and current smoking status are predictive of COPD mortality. Not all smokers develop clinically significant COPD, which suggests that genetic factors must modify each individual’s risk. Although it is unclear what percentage of smokers develop the disease, the commonly cited figure of 15-20% is likely an underestimate because COPD is both under-diagnosed and under-appreciated.
Passive exposure to cigarette smoke (also known as environmental tobacco smoke or ETS) may also contribute to respiratory symptoms and COPD by increasing the lungs’ total burden of inhaled particles and gases2,42,43. Smoking during pregnancy may also pose a risk for the fetus, by affecting lung growth and development in utero and possibly the priming of the immune system32,44.
Figure 3.3 Interaction of Smoking and Occupational Exposures
£ No ¢ Yes
Occupational dusts and chemicals (vapors, irritants, and fumes) can also cause COPD when the exposures are sufficiently intense or prolonged, such as those experienced by miners in many countries. These exposures can both cause COPD independently of cigarette smoking and increase the risk in the presence of concurrent cigarette smoking 41. Exposure to coal dust alone in sufficient doses can produce airflow limitation45,46.
Exposure to particulate matter, irritants, organic dusts, and sensitizing agents can cause an increase in airway hyperresponsiveness47, especially in airways already damaged by other occupational exposures, cigarette smoke, or asthma. There is some evidence from community studies that a combination of dust exposure and gas or fume exposure may have an additive effect on the risk of COPD48-50.
High levels of urban air pollution are harmful to individuals with existing heart or lung disease. The role of outdoor air pollution in causing COPD is unclear, but appears to be small when compared with that of cigarette smoking. The relative effect of short-term, high peak exposures and long-term, low-level exposures is a question yet to be resolved.
Over the past two decades, air pollution in most cities in developed countries has decreased appreciably. In contrast, air pollution has increased markedly in many cities in developing countries. Although it is not clear which specific elements of ambient air pollution are harmful, there is some evidence that particles found in polluted air will add to a person’s total inhaled burden. Indoor air pollution from biomass fuel has been implicated as a risk factor for the development of COPD. This exposure is greatest in regions where biomass fuel is used for cooking and heating in poorly vented dwellings, leading to high levels of particulate matter in indoor air51-61.
A history of severe childhood infection has been associated with reduced lung function and increased respiratory symptoms in adulthood32. There are several possible explanations for this association (which are not mutually exclusive). There may be an increased diagnosis of severe infections in children who have underlying airway hyperresponsiveness, itself considered a risk factor for COPD. Viral infections may be related to another factor, such as birth weight, that is related to COPD.
HIV infection has been shown to accelerate the onset of smoking-induced emphysema; HIV-induced pulmonary inflammation may play a role in this process62-66.
There is evidence that the risk of developing COPD is inversely related to socioeconomic status65. It is not clear, however, whether this pattern reflects exposures to indoor and outdoor air pollutants, crowding, poor nutrition, or other factors that are related to low socioeconomic status60,65.
1. Feinleib M, Rosenberg HM, Collins JG, Delozier JE, Pokras R, Chevarley FM. Trends in COPD morbidity and mortality in the United States. Am Rev Respir Dis 1989; 140:S9-18.
2. Buist AS, Vollmer WM. Smoking and other risk factors. In: Murray JF, Nadel JA, eds. Textbook of respiratory medicine. Philadelphia: WB Saunders Co; 1994. p. 1259-87.
3. Thom TJ. International comparisons in COPD mortality. Am Rev Respir Dis 1989; 140:S27-34.
4. Xu X, Weiss ST, Rijcken B, Schouten JP. Smoking, changes in smoking habits, and rate of decline in FEV1: new insight into gender differences. Eur Respir J 1994; 7:1056-61.
5. National Center for Health Statistics. Current estimates from the National Health Interview Survey, United States, 1995. Washington, DC: Department of Health and Human Services, Public Health Service, Vital and Health Statistics; 1995. Publication No. 96-1527.
6. National Heart, Lung, and Blood Institute. Morbidity & mortality: chartbook on cardiovascular, lung, and blood diseases. Bethesda, MD: US Department of Health and Human Services, Public Health Service, National Institutes of Health; 1998. Available from: URL: www.nhlbi.nih.gov/nhlbi/seiin/other/cht-book/htm
7. Anthonisen NR, Connett JE, Kiley JP, Altose MD, Bailey WC, Buist AS, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA 1994; 272:1497-505.
8. Wilson DO, Rogers RM, Wright EC, Anthonisen NR. Body weight in chronic obstructive pulmonary disease. The National Institutes of Health Intermittent Positive-Pressure Breathing Trial. Am Rev Respir Dis 1989; 139:1435-8.
9. Sahebjami H, Vassallo CL. Influence of starvation on enzyme-induced emphysema. J Appl Physiol 1980; 48:284-8.
10. Silverman EK, Speizer FE. Risk factors for the development of chronic obstructive pulmonary disease. Med Clin North Am 1996; 80:501-22.
11. Chen Y. Genetics and pulmonary medicine.10: Genetic epidemiology of pulmonary function. Thorax 1999; 54:818-24.
12. Laurell CB, Eriksson S. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132-40.
13. Hubbard RC, Crystal RG. Antiproteases. In: Crystal RB, West JB, Barnes PJ, Cherniack NS, Weibel ER, eds. The lung: scientific foundations. New York: Raven Press; 1991. p. 1775-87.
14. McElvaney NG, Crystal RG. Inherited susceptibility of the lung to proteolytic injury. In: Crystal RG, West JB, Weibel ER, Barnes PJ, eds. The lung: scientific foundations. Philadelphia: Lippincott-Raven; 1997. p. 2537-53.
15. Khoury MJ, Beaty TH, Newill CA, Bryant S, Cohen BH. Genetic-environmental interactions in chronic airways obstruction. Int J Epidemiol 1986; 15:65-72.
16. Cohen BH, Bias WB, Chase GA, Diamond EL, Graves CG, Levy DA, et al. Is ABH nonsecretor status a risk factor for obstructive lung disease? Am J Epidemiol 1980; 111:285-91.
17. Smith CA, Harrison DJ. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet 1997; 350:630-3.
18. Harrison DJ, Cantlay AM, Rae F, Lamb D, Smith CA. Frequency of glutathione S-transferase M1 deletion in smokers with emphysema and lung cancer. Hum Exp Toxicol 1997; 16:356-60.
19. Faber JP, Poller W, Olek K, Baumann U, Carlson J, Lindmark B, et al. The molecular basis of alpha 1-antichymotrypsin deficiency in a heterozygote with liver and lung disease. J Hepatol 1993; 18:313-21.
20. Schellenberg D, Pare PD, Weir TD, Spinelli JJ, Walker BA, Sandford AJ. Vitamin D binding protein variants and the risk of COPD. Am J Respir Crit Care Med 1998; 157:957-61.
21. Huang SL, Su CH, Chang SC. Tumor necrosis factor-alpha gene polymorphism in chronic bronchitis. Am J Respir Crit Care Med 1997; 156:1436-9.
22. Siafakas NM, Tzortzaki EG, Sourvinos G, Bouros D, Tzanakis N, Kafatos A, et al. Microsatellite DNA instability in COPD. Chest 1999; 116:47-51.
23. Orie NGM, Sluiter HJ, De Vreis K, Tammerling K, Wikop J. The host factor in bronchitis. In: Orie NGM, Sluiter HJ, eds. Bronchitis, an international symposium. Assen, Netherlands: Royal Vangorcum; 1961. p. 43-59.
24. Peat JK, Woolcock AJ, Cullen K. Rate of decline of lung function in subjects with asthma. Eur J Respir Dis 1987; 70:171-9.
25. Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194-200.
26. Tashkin DP, Altose MD, Connett JE, Kanner RE, Lee WW, Wise RA. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group. Am J Respir Crit Care Med 1996; 153:1802-11.
27. Morgan WJ. Maternal smoking and infant lung function. Further evidence for an in utero effect [editorial; comment]. Am J Respir Crit Care Med 1998; 158:689-90.
28. Hagstrom B, Nyberg P, Nilsson PM. Asthma in adult life - is there an association with birth weight? Scand J Prim Healthcare1998; 16:117-20.
29. Svanes C, Omenaas E, Heuch JM, Irgens LM, Gulsvik A. Birth characteristics and asthma symptoms in young adults: results from a population-based cohort study in Norway. Eur Respir J 1998; 12:1366-70.
30. Todisco T, de Benedictis FM, Iannacci L, Baglioni S, Eslami A, Todisco E, et al. Mild prematurity and respiratory functions. Eur J Pediatr 1993; 152:55-8.
31. Stein CE, Kumaran K, Fall CH, Shaheen SO, Osmond C, Barker DJ. Relation of fetal growth to adult lung function in South India. Thorax 1997; 52:895-9.
32. Tager IB, Segal MR, Speizer FE, Weiss ST. The natural history of forced expiratory volumes. Effect of cigarette smoking and respiratory symptoms. Am Rev Respir Dis 1988; 138:837-49.
33. US Surgeon General. The health consequences of smoking: chronic obstructive pulmonary disease. Washington, D.C: US Department of Health and Human Services; 1984. Publication No. 84-50205.
34. Higgins MW, Thom T. Incidence, prevalence, and mortality: intra- and inter-country differences. In: Hensley M, Saunders N, eds. Clinical epidemiology of chronic obstructive pulmonary disease. New York: Marcel Dekker; 1989. p. 23-43.
35. Sherrill DL, Lebowitz MD, Burrows B. Epidemiology of chronic obstructive pulmonary disease. Clin Chest Med 1990; 11:375-87.
36. Auerbach O, Hammond EC, Garfinkel L, Benante C. Relation of smoking and age to emphysema. Whole-lung section study. N Engl J Med 1972; 286:853-7.
37. Burrows B, Knudson RJ, Cline MG, Lebowitz MD. Quantitative relationships between cigarette smoking and ventilatory function. Am Rev Respir Dis 1977; 115:195-205.
38. Lebowitz MD, Burrows B. Quantitative relationships between cigarette smoking and chronic productive cough. Int J Epidemiol 1977; 6:107-13.
39. Higgins MW, Keller JB, Becker M, Howatt W, Landis JR, Rotman H, et al. An index of risk for obstructive airways disease. Am Rev Respir Dis 1982; 125:144-51.
40. Becklake MR. Occupational exposures: evidence for a causal association with chronic obstructive pulmonary disease. Am Rev Respir Dis 1989; 140:S85-91.
41. Kauffmann F, Drouet D, Lellouch J, Brille D. Twelve years spirometric changes among Paris area workers. Int J Epidemiol 1979; 8:201-12.
42. Leuenberger P, Schwartz J, Ackermann-Liebrich U, Blaser K, Bolognini G, Bongard JP, et al. Passive smoking exposure in adults and chronic respiratory symptoms (SAPALDIA Study). Swiss Study on Air Pollution and Lung Diseases in Adults, SAPALDIA Team. Am J Respir Crit Care Med 1994; 150:1222-8.
43. Dayal HH, Khuder S, Sharrar R, Trieff N. Passive smoking in obstructive respiratory disease in an industrialized urban population. Environ Res 1994; 65:161-71.
44. Holt PG. Immune and inflammatory function in cigarette smokers. Thorax 1987; 42:241-9.
45. US Centers for Disease Control and Prevention. Criteria for a recommended standard: occupational exposure to respirable coal mine dust. Morgantown, WV: National Institute of Occupational Safety and Health; 1995. Publication No. 95-106.
46. Heppleston AG. Prevalence and pathogenesis of pneumoconiosis in coal workers. Environ Health Perspect 1988; 78:159-70.
47. Niewoehner DE. Anatomic and pathophysiological correlations in COPD. In: Baum GL, Crapo JD, Celli BR, Karlinsky JB, eds. Textbook of pulmonary diseases. Philadelphia: Lippincott-Raven; 1998. p. 823-42.
48. Bakke S, Baste V, Hanoa R, Gulsvik A. Prevalence of obstructive lung disease in a general population: relation to occupational title and exposure to some airborne agents. Thorax 1991; 46:863-70.
49. Humerfelt S, Gulsvik A, Skjaerven R, Nilssen S, Kvale G, Sulheim O, et al. Decline in FEV1 and airflow limitation related to occupational exposures in men of an urban community. Eur Respir J 1993; 6:1095-103.
50. Humerfelt S, Eide GE, Gulsvik A. Association of years of occupational quartz exposure with spirometric airflow limitation in Norwegian men aged 30-46 years. Thorax 1998; 53:649-55.
51. Chen JC, Mannino MD. Worldwide epidemiology of chronic obstructive pulmonary disease. Current Opinion in Pulmonary Medicine 1999; 5:93-9.
52. Perez-Padilla R, Regalado U, Vedal S, Pare P, Chapela R, Sansores R, et al. Exposure to biomass smoke and chronic airway disease in Mexican women. Am J Respir Crit Care Med 1996; 154:701-6.
53. Dossing M, Khan J, al-Rabiah F. Risk factors for chronic obstructive lung disease in Saudi Arabia. Respiratory Med 1994; 88:519-22.
54. Behera D, Jindal SK. Respiratory symptoms in Indian women using domestic cooking fuels. Chest 1991; 100:385-8.
55. Amoli K. Bronchopulmonary disease in Iranian housewives chronically exposed to indoor smoke. Eur Respir J 1998; 11:659-63.
56. Dennis R, Maldonado D, Norman S, Baena E, Martinez G. Woodsmoke exposure and risk for obstructive airways disease among women. Chest 1996; 109:115-9.
57. Pandey MR. Prevalence of chronic bronchitis in a rural community of the Hill Region of Nepal. Thorax 1984; 39:331-6.
58. Pandey MR. Domestic smoke pollution and chronic bronchitis in a rural community of the Hill Region of Nepal. Thorax 1984; 39:337-9.
59. Samet JM, Marbury M, Spengler J. Health effects and sources of indoor air pollution. Am Rev Respir Dis 1987; 136:1486-508.
60. Tao X, Hong CJ, Yu S, Chen B, Zhu H, Yang M. Priority among air pollution factors for preventing chronic obstructive pulmonary disease in Shanghai. Sci Total Environ 1992; 127:57-67.
61. Smith KR. National burden of disease in India from indoor air pollution. Proc Natl Acad Sci USA 2000; 97:13286-93.
62. Diaz PT, Clanton TL, Pacht ER. Emphysema-like pulmonary disease associated with human immunodeficiency virus infection. Ann Intern Med 1992; 116:124-8.
63. Diaz PT, King MA, Pacht ER, Wewers MD, Gadek JE, Nagaraja HN, et al. Increased susceptibility to pulmonary emphysema among HIV-seropositive smokers. Ann Intern Med 2000; 132:369-72.
64. Diaz PT, King MA, Pacht ER, Wewers MD, Gadek JE, Neal D, et al. The pathophysiology of pulmonary diffusion impairment in human immunodeficiency virus infection. Am J Respir Crit Care Med 1999; 160:272-7.
65. Prescott E, Lange P, Vestbo J. Socioeconomic status, lung function and admission to hospital for COPD: results from the Copenhagen City Heart Study. Eur Respir J 1999; 13:1109-14.
66. Strachan DP. Epidemiology: a British perspective. In: Calverley PMA, Pride NB, eds. Chronic obstructive pulmonary disease. London: Chapman and Hall; 1995. p. 47
| Course |